MDBenchmark: Benchmark molecular dynamics simulations¶




MDBenchmark — quickly generate, start and analyze benchmarks for your molecular dynamics simulations.
MDBenchmark is a tool to squeeze the maximum out of your limited computing resources. It tries to make it as easy as possible to set up systems on varying numbers of nodes and compare their performances to each other.
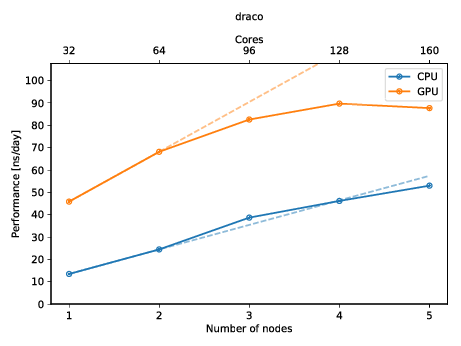
You can also create a plot to get a quick overview of the possible performance (and show of to your friends)! The plot below shows the performance of an molecular dynamics system on up to five nodes with and without GPUs.
Quick start¶
Follow the next two paragraphs to get a quick start. Extended usage guides can
be found below. You can install mdbenchmark with your favorite Python
package manager. Afterwards you are ready to use mdbenchmark.
Install¶
If you are familiar with the usual way of installing python packages, just use
pip:
pip install mdbenchmark
Anaconda users can install via conda:
conda install -c conda-forge mdbenchmark
Cutting-edge users may prefer pipenv:
pipenv install mdbenchmark
Usage¶
Now that the package is installed, you can generate benchmarks for your system.
Assuming you want to benchmark a GROMACS 2018.2 simulation on up to 5 nodes,
with the TPR file called md.tpr, run the following command:
mdbenchmark generate -n md --module gromacs/2018.2 --max-nodes 5
After generation benchmarks can be submitted:
mdbenchmark submit
Now, you can also monitor the status of your benchmark with mdbenchmark.
This will show you the performance of all runs that have finished:
mdbenchmark analyze
Plotting of the current results can be achieved with mdbenchmark analyze
--plot.
Usage reference¶
mdbenchmark¶
Generate, run and analyze benchmarks of GROMACS simulations.
mdbenchmark [OPTIONS] COMMAND [ARGS]...
Options
-
--version¶ Show the version and exit.
analyze¶
Analyze finished benchmarks.
mdbenchmark analyze [OPTIONS]
Options
-
-d,--directory<directory>¶ Path in which to look for benchmarks. [default: .]
-
-p,--plot¶ Generate a plot of finished benchmarks.
-
--ncores<ncores>¶ Number of cores per node. If not given it will be parsed from the benchmarks log file.
-
-o,--output-name<output_name>¶ Name of the output .csv file.
generate¶
Generate benchmarks simulations from the CLI.
mdbenchmark generate [OPTIONS]
Options
-
-n,--name<name>¶ Name of input files. All files must have the same base name.
-
-g,--gpu¶ Use GPUs for benchmark. [default: False]
-
-m,--module<module>¶ Name of the MD engine module to use.
-
--host<host>¶ Name of the job template.
-
--min-nodes<min_nodes>¶ Minimal number of nodes to request. [default: 1]
-
--max-nodes<max_nodes>¶ Maximal number of nodes to request. [default: 5]
-
--time<time>¶ Run time for benchmark in minutes. [default: 15]
-
--list-hosts¶ Show available job templates.
-
--skip-validation¶ Skip the validation of module names.
submit¶
Submit benchmarks to queuing system.
benchmarks are searched recursively starting from the directory specified in –directory.
Checks whether benchmark folders were already generated, exits otherwise. Only runs benchmarks that were not already started. Can be overwritten with –force.
mdbenchmark submit [OPTIONS]
Options
-
-d,--directory<directory>¶ Path in which to look for benchmarks. [default: .]
-
-f,--force¶ Resubmit all benchmarks and delete all previous results.